Eredeti cikk dátuma: 2020. április 23.
Eredeti cikk címe: Patient-derived mutations impact pathogenicity of SARS-CoV-2
Eredeti cikk szerzői: Hangping Yao, Xiangyun Lu, Qiong Chen, Kaijin Xu, Yu Chen, Linfang Cheng, Fumin Liu, Zhigang Wu, Haibo Wu, Changzhong Jin, Min Zheng, Nanping Wu, Chao Jiang, Lanjuan Li
Eredeti cikk elérhetősége: https://www.medrxiv.org/content/10.1101/2020.04.14.20060160v2.full.pdf
Eredeti cikk státusza:
Fordító(k): dr. Serly Juliannana
Lektor(ok): dr. Szabó Edit
Nyelvi lektor(ok): Rét Anna
Szerkesztő(k): Novák Zsuzsanna
Figyelem! Az oldalon megjelenő cikkek esetenként politikai jellegű megnyilvánulásokat is tartalmazhatnak. Ezek nem tekinthetők a fordítócsoport politikai állásfoglalásának, kizárólag az eredeti cikk írójának véleményét tükrözik. Fordítócsoportunk szigorúan politikamentes, a cikkekben esetlegesen fellelhető politikai tartalommal kapcsolatosan semmiféle felelősséget nem vállal, diskurzust, vitát, bizonyítást vagy cáfolatot nem tesz közzé.
Az oldalon található információk nem helyettesítik a szakemberrel történő személyes konzultációt és kivizsgálást, ezért kérjük, minden esetben forduljon szakorvoshoz!
Összefoglalás (130 szó)
A súlyos akut légzőszervi szindrómát okozó koronavírus (SARS-CoV-2) az egész világon elterjedt, több mint 1 300 000 beteget diagnosztizáltak, és a halálos áldozatok száma eléri a 70 000 főt. Nemrégiben megjelent genomikai tanulmányokból származó adatok szerint jelentős lehet az egynukleotid-variánsok (SNV-k) száma. Jelenleg nem kapcsolnak egyetlen mutációt sem közvetlenül a víruspatogenitás funkcionális változásaihoz. Ebben a publikációban 11 betegből származó vírusizolátum funkcionális jellemzőit írjuk le. Sokféle mutációt figyeltünk meg ezekben a vírusizolátumokban, többek között 6 különböző mutációt a glikoproteintüskén (S-fehérjén), és ezek között két olyan különböző SNV-t találtunk, amelyek ugyanahhoz a misszensz mutációhoz vezetnek. Fontos megjegyezni, hogy ezek a vírusizolátumok szignifikáns változatosságot mutatnak citopátiás hatásukban és a vírusterhelésben, közel 270-szeres különbséget mértünk, amikor Vero-E6-sejteket fertőztünk. Ezért közvetlen bizonyítékot szolgáltatunk arra, hogy a SARS-CoV-2-mutációk között vannak olyanok, amelyek alapvetően képesek megváltoztatni a vírus patogenitását.
Bevezetés
A súlyos akut légzőszervi szindrómát okozó koronavírus 2 (SARS-CoV-2, korábbi hivatkozásokban 2019-nCoV), amelyet a jelenlegi járványkitörésre jellemző atípusos tüdőgyulladással hoznak összefüggésbe, a közép-kínai Vuhanból indult világjárvány oka, annak ellenére, hogy Kína kiterjedt, rendszerszintű erőfeszítéseket tett a kitörés megfékezésére. 2020. április 7-éig a SARS-CoV-2 több mint 1,3 millió embert fertőzött meg, és a halálos áldozatok száma eléri a 70 000 főt. A számok még mindig gyorsan emelkednek. A SARS-CoV-2 becsült inkubációs ideje (átlag 5,1 nap; mérési tartomány 4,5–5,8 nap) (Lauer et al., 2020) egybecseng más humán koronavírusokéval, mint a SARS (átlag 5 nap; mérési tartomány 2–14 nap) (Varia et al., 2003) és a MERS (átlag 5–7 nap; mérési tartomány 2–14 nap) (Virlogeux et al., 2016). A SARS-CoV-2 reprodukciós száma valószínűleg 1,4–6,5, átlag 3,3 (Liu et al., 2020), ami némileg magasabb, mint a SARS esetében tapasztalt 2–5 (Bauch et al., 2005; Lipsitch et al., 2003) és a MERS-nél mért 2,7–3,9 (Lin et al., 2018). A SARS-CoV-2-vel fertőzött betegek több mint fele nem lázasodott be, mielőtt kórházba került (Guan et al., 2020). Meglepő, hogy a 2019-es koronavírus-betegséget (COVID19) tünetmentes hordozók is terjeszthetik, akik soha nem lázasodnak be, nem mutatnak emésztőrendszeri vagy légzőszervi tüneteket, és normális a mellkasi CT-jük is (Bai et al., 2020; Hu et al., 2020), ami nagyon nehéz kihívássá teszi a COVID19 terjedésének megállítását. A SARS-CoV-2 továbbá intakt és fertőzőképes marad több órán át aeroszolban, és akár 7 napig is a felületeken (van Doremalen et al., 2020). Habár több in vitrotanulmány vagy klinikai vizsgálat folyik inhibitorokkal vagy gyógyszerekkel kapcsolatban, eddig még nem találtak hatékony gyógymódot vagy oltóanyagot (Cao et al., 2020; Hoffmann et al., 2020; Wang et al., 2020). Az Egészségügyi Világszervezet (WHO) 2020. január 30-án nemzetközi jelentőségű közegészségügyi vészhelyzetté (PHEIC) nyilvánította a COVID19-et, és felhívta a figyelmet 2020. február 28-án arra a fenyegetettségre, hogy a SARS-CoV-2-fertőzés világjárvánnyá válásának valószínűsége „nagyon magas”.
A SARS-CoV-2 a burkos RNS bétakoronavírusok hetedik képviselője (Sarbecovirus alnemzetség) (Zhu et al., 2020); ezek közül a SARS-CoV-2, a SARS-CoV és a MERS-CoV pusztító betegséget képes okozni, míg a HKU1, az NL63, az OC43 és a 229E vírusok enyhe tüneteket okoznak (Corman et al., 2018). Eddig nem találtak rekombináns eseményre utaló jelet (Yu, 2020), habár ez lehet részben annak a ténynek köszönhető, hogy a legtöbb vírusizolátumot short-read-es (rövid leolvasási) platformokkal szekvenálták. A transzmembrán tüske(S)-glikoprotein irányítja a vírusbejutást a gazdasejtbe a vírus felszínéről kiemelkedő homotrimerek segítségével. Az S-proteinnek két doménje van: Az S1 felelős a gazdasejt receptorához kötődésért, az S2 pedig a vírus és a sejtmembrán fúziójáért (Tortorici és Veesler, 2019). A SARS-CoV-2 és a SARS-CoV is az angiotenzinkonvertáló enzim 2 (ACE2) receptorát használja a célsejtbe jutáshoz (Walls et al., 2020). Az ACE2 receptora a humán nazális epitélsejteken, valamint a tüdő, a spermatogónium, a leydig, a sertoli, a gyomor, a vékonybél és a végbél epitélsejtjein expresszálódik (Wang and Xu, 2020; Xiao et al., 2020; Zhao et al., 2020). Az S-fehérje receptorkötő doménje (RBD) mutatja a legnagyobb genetikai változatosságot a bétakoronavírus csoportban (Wu et al., 2020; Zhou et al., 2020), és az S-fehérje lehet a pozitív szelekció alanya (Lv et al., 2020). A SARS-CoV-2 variabilitása ellenére egy kérdés még megválaszolatlan marad, hogy vajon ezeknek a mutációknak van-e bármilyen tényleges funkcionális hatása a SARS-CoV-2 patogenitására. Nagyon fontos megértenünk a vírus fertőzési mechanizmusait, és meghatározni a gyógyszer- és vakcinafejlesztési stratégiákat a világjárvány következő szakaszára való felkészülés során.
Emiatt 11 SARS-CoV-2-betegből származó vírusizolátumot jellemeztünk, a betegeket a Csöcsiangi Egyetemi Kórházhoz tartozó társintézményekbe vették fel a kínai Hangzhouban, amely 757 km-re található Vuhantól (ld. Anyagok és módszerek). A 11 vírusizolátum szupermély szekvenálása során a Novaseq 6000 platformon 1-5 mutációt azonosítottunk a vírusizolátumok kódoló szekvenciáin. Kevert víruspopulációkat is megfigyeltünk (amelyek a kvázi fajokat képviselik). Vero-E6-sejteket fertőztünk 11 vírusizolátummal és kvantitatívan határoztuk meg a vírusterhelésüket 1, 2, 4, 8, 24 és 48 órával a fertőzés után, valamint meghatároztuk a citopátiás hatást (CPE) is 48 és 72 órával a fertőzést követően. Az eredményeink arra utalnak, hogy a megfigyelt mutációknak közvetlen hatása lehet a vírusterhelésre és a CPE-re a Vero-E6-sejtek fertőzésekor; legalább 270-szeres különbség figyelhető meg a két véglet között. Ez az eredmény azt sugallja, hogy a vizsgálatunkban megfigyelt mutációk, és valószínűleg a világon összegyűjtött valamennyi vírusizolátum mutációja jelentősen befolyásolhatja a SARS-CoV-2 patogenitását.
Eredmények
A betegek epidemiológiai előzményeinek összefoglalása
Az ebbe a vizsgálatba bevont 11 betegtől származó mintát a COVID19 korai kínai kitörése során gyűjtöttük, 2020. január 22-e és 2020. február 4-e között (1. táblázat). 11-ből 10 betegnél egyértelmű a kapcsolat Vuhan városával, ahol a SARS-CoV-2-t eredetileg azonosították. 11-ből 5 beteg Vuhanban dolgozott vagy odautazott, mielőtt diagnosztizálták volna, és további 5 beteg közeli kontaktusba került olyanokkal, akik Vuhanban éltek, a maradék egy beteg pedig egy elhunyt COVID19-beteggel állt kapcsolatban. Megjegyezzük, hogy a ZJU-4, -5 és -9 számú betegek ugyanazon az üzleti konferencián vettek részt, ahol néhány vuhani kollégájuk is megjelent. Ezek a betegek ezért 1. vagy 2. generációs betegnek tekinthetők a vírus szempontjából az epidemiológiai előzményt figyelembe véve. A 11 beteg között 8 férfi és 3 nő volt, a 4 hónapostól a 71 évesig. Nem volt más szelekciós kritérium e betegek kiválasztására, mint hogy mindannyian a Csöcsiangi Egyetemhez tartozó hangzhoui kórházakba kerültek. Egy beteg kivételével középsúlyos vagy súlyosabb tüneteik voltak. 3 betegnél nem állt fenn társbetegség, és egy beteg igényelt intenzív osztályos ellátást. Szerencsére minden beteg felépült, mire ezt a cikket elkezdtük megírni.
| Azonosítószám | Nem | Életkor | Minta | Minta mintavétel dátuma | Vírusizoláció dátuma | Epidemiológia | Vírusgen. | A jelentkező tünetek | Súlyosság | Vérnyomás | ITO | Tünetek jelentkezésének dátuma | Felvétel dátuma | Elbocsátás |
| ZJU_1 | F | 36 | Köpet | 01.25. | 01.29. | Kontaktus vuhani személlyel | 2 | Láz | Középsúlyos | 0 | 0 | 01.23. | 01.25. | 02.23. |
| ZJU_2 | F | 31 | Köpet | 01.26. | 01.29. | Vuhanban élt | 1 | Láz | Súlyos | 0 | 0 | 01.23. | 01.24. | 02.23. |
| ZJU_3 | F | 32 | Köpet | 01.25. | 01.30. | Vuhanba utazott | 2 | Láz | Súlyos | 0 | 0 | 01.18. | 01.19. | 02.12. |
| ZJU_4 | F | 34 | Köpet | 01.24. | 01.28. | Konferencián vett részt vuhani kollégákkal | 2 | Láz, fáradékonyság | Középsúlyos | 0 | 0 | 01.17. | 01.21. | 02.09. |
| ZJU_5 | N | 25 | Köpet | 01.22. | 01.26. | Konferencián vett részt vuhani kollégákkal | 2 | Láz | Súlyos | 0 | 0 | 01.21. | 01.22. | 02.09. |
| ZJU_6 | F | 71 | Köpet | 02.02. | 02.06. | Kontaktus COVID19-beteg vuhani személlyel. | 2 vagy 3 | Láz, fáradékonyság | Súlyos | 1 | 0 | 01.22. | 01.26. | 02.19. |
| ZJU_7 | N | 4 hó | Orrgaratból vett minta | 02.03. | 02.08. | Kontaktus vuhani személlyel | 2 | Láz | Enyhe | 0 | 0 | 01.29. | 01.29. | 02.20. |
| ZJU_8 | F | 53 | Köpet | 01.26. | 01.30. | Vuhanban élt | 1 | Láz, fáradékonyság | Kritikus | 1 | 1 | 01.17. | 01.22. | 02.19. |
| ZJU_9 | F | 30 | Széklet | 01.28. | 02.07. | Konferencián vett részt vuhani kollégákkal | 2 | Láz | Súlyos | 0 | 0 | 01.18. | 01.21. | 02.05. |
| ZJU_10 | N | 38 | Széklet | 02.03. | 02.08. | Vuhanban élt | 1 | Láz | Súlyos | 0 | 0 | 01.19. | 01.27. | 02.12. |
| ZJU_11 | F | 62 | Széklet | 02.04. | 02.09. | Vuhanban élt | 1 | Köhögés | Súlyos | 1 | 0 | 01.19. | 01.26. | 03.15. |
A betegekből származó vírusizolátumokban az ultramély szekvenálás különböző mutációkat fedett fel.
A 11 vírusizolátum mutációs spektrumának meghatározásához Illumina Novaseq 6000 platformon végeztük el az izolált vírusgenomi RNS ultramély szekvenálását, amivel tisztítás után átlagosan 245 millió readhez jutottunk/67,16 Gb mintánként (S1 táblázat; az átlagos lefedettség legalább kétmilliószoros volt). Ez a kivételes mélység részben annak köszönhető, hogy a SARS-CoV-2-nek kicsi a genomja, ami lehetővé teszi, hogy a mutációkat nagy bizonyossággal azonosítani lehessen. Azokban az esetekben, amikor a víruspopulációk nem voltak homogének, a mélység segített az alacsony frekvenciájú allélek azonosításában.
Összesen 33 mutációt azonosítottunk (beleértve a kevert populációkban talált 10 mutációt is), és ezek közül 19 új mutáció, összehasonlítva a GISAID-on 2020. március 24-én elérhető 1111 genomi szekvenciával (1., S1 és S2 ábra). A G11083T és G26144T mutációkat a ZJU-1 betegben azonosítottuk, és mindkét mutációról ismert, hogy egy nagy víruscsoport alapító mutációinak számítanak (Capobianchi et al., 2020). A C8782T és T28144C mutációkat megtaláltuk négyből két vírusizolátumban, a ZJU-2 és a ZJU-8 számúban, és mindkét mutációról ismert, hogy ezek egy másik nagy víruscsoport alapító mutációi (Capobianchi et al., 2020). Érdekes, hogy a T22303G mutációt öt vírusizolátumban azonosítottuk (ZJU-2, -5, -9, -10 és -11), és a ZJU-5 és ZJU-9 számú beteg ugyanannak a fertőzési forrásnak lehetett kitéve az üzleti konferencia alatt (1. táblázat). Korábban csak egy vírusizolátumot azonosítottak Ausztráliában a T22303G mutációval. Feltűnő, hogy a ZJU-5 és ZJU-9 számú beteggel ugyanazon konferencián részt vevő ZJU-4 számú beteg vírusizolátumában egy új mutációt, az A22301C-t azonosítottunk, amely ugyanazt a misszensz mutációt okozza fehérjeszinten (S247R az S-fehérjében), mint a T22303G, ahol az első pozíció helyett a harmadikban történt a mutáció ugyanabban a kodonban. E két egynukleotid-variánssal kapcsolatos megfigyelés csak véletlen lehet, ennek ellenére elég váratlan. Végül a ZJU-11 számú izolátumban 4 mutációt találtunk az ORF7b génben, amelyből 3 egymás után található, és két mutációt okoz fehérjeszinten. A di- és trinukleotid-mutációk természetesen ritkábbak, mint az SNV, de nem extrém ritkák, a korábban prokariótákban vizsgált mutációakkumulációs tanulmányok szerint (Lynch, 2007).
Fontos megjegyezni, hogy bár a szekvenciaadatokat a GISAID tárolja, ami nagyon hasznos a vírus betegek közti eltéréseinek nyomon követéséhez, még mindig nem tudunk túl sokat az adott személyen belül bekövetkező virális evolúciós dinamikáról. Például a ZJU-4 és a ZJU-10 mintákban két különböző helyen lokalizálódó allél nagyon hasonló gyakorisági eloszlást mutatott, ami azt jelzi, hogy ez a két hely valószínűleg kapcsolt, és ez legalább két haplotípust jelent a víruspopulációban. És ahogy ebben a vizsgálatban kiderült, az azonosított mutációk közül 6-ot figyelmen kívül hagytunk volna, ha a konszenzusszekvencia-analízist használjuk. Mindent egybevetve, annak ellenére, hogy csak 11 betegből származó izolátumot vizsgáltunk ebben a tanulmányban, megfigyelhettük a bőséges mutációs diverzitást, amely több olyan nagy vírusklaszter számos alapító mutációját tartalmazza, amelyek jelenleg az egész világon elterjedtek. Ez a diverz mutációs spektrum egybecseng a viszonylag korai mintavétellel, és a Vuhan városától mért viszonylagos közelséggel, ahol az első vírustörzset azonosították. A vírus teljes mutációs diverzitása Vuhan városában a járvány elejéről még napjainkig sem ismert, aminek oka a korlátozott mintavétel (Lu et al., 2020; Zhou et al., 2020).

1. ábra A 11 vírusizolátumban azonosított mutációk összegzése. A vírusgenom minden ORF-ét az NCBI NC_045512.2 jelölésmódja szerint adták meg. Ha egy mutációt kevert populációban figyeltek meg, akkor a két leggyakoribb allél százalékos arányait adták meg. Az aminosavszinten bekövetkező változásokat is megadták, ahol felmerült. A kék szín a közlemény írásának idején újnak számító mutációt jelöli.
A betegekből származó vírusizolátumok filogenetikai elemzéséből kiderülnek a szerteágazó evolúciós előzmények
Hogy megértsük a 11 vírusizolátum filogenetikai kontextusát az elérhető SARS-CoV-2 szekvenálási adataival összefüggésben, 725 jó minőségű és nagy lefedettségű SARS-CoV-2 genomot szereztünk be a GISAID-tól (letöltve 2020. március 21-én), beleértve a Yunnan RaTG13 vírustörzset és a Guangdong tobzoska vírustörzset is mint külső referenciát (outgroup). A 736 genomi szekvenciát MAFFT-tal (ld. Anyagok és módszerek) illesztettük egymáshoz, és trimAL-lal trimmeltük a teljes illesztést (ld. Anyagok és módszerek), hogy eltávolítsuk az illesztés hamis részeit. Az IQ-TREE-t használtuk (ld. Anyagok és módszerek), hogy a 736 vírusszekvencia egy 835 parszimóniainformatív karaktert tartalmazó illesztéséből maximum likelihood törzsfát becsüljünk 1000 bootstrap ismétléssel (2A ábra). A létrehozott törzsfa nagymértékben konzisztens a GISAID-on frissített filogenetikai elemzéssel (S3 ábra). Szeretnénk hangsúlyozni, hogy a COVID19-helyzettel kapcsolatos gyors lépéseknek köszönhetően tízesével, vagy inkább százasával állnak rendelkezésre új szekvenciák, amelyeket nap mint nap feltöltenek a GISAID-ra. Emiatt új megfigyeléseket lehet majd generálni, ha több adat áll rendelkezésre.

2. ábra A betegekből származó SARS-CoV-2-izolátumok jellemzése. (A) 11 vírusizolátum filogenetikai analízise a GISAID-ról letöltött 725 SARS-CoV-2 szekvencia segítségével. A bootstrap-eljárás során 1000 ismétléssel becsült maximum likelihood törzsfát a 11 vírusizolátum filogenetikai kontextusának szemléltetésére hoztuk létre. A nagyobb és a kisebb klasztereket színkódoztuk; a színmagyarázat az ábra bal felső részén található. Minden ZJU-mintához a zöld színt rendeltük hozzá. Az ág vastagsága a bootstrap-támogatottsággal arányos. (B) A fluoreszcensen jelölt virális S-fehérje azt mutatja meg, hogy az izolált SARS-CoV-2 virionok (zöld) a Vero-E6-sejtek külső részéhez kötődnek (a DNS kéken festődik) a bejutás előtt. Léptéksáv 50 µm. (C) Az izolált SARS-CoV-2 virionok tipikus TEM-képe, ahol a nyilak az S-fehérjékből (tüske) álló jellegzetes „koronákra” mutatnak. Léptéksáv 100 nm.
Elég kevés alapító mutációcsoportot figyeltünk meg. Konkrétan a filogenetikai elemzésünk során a következő három legnagyobb klasztert vesszük figyelembe: 1. A C241T (csendes), a C14408T (csendes) és az A23403G (D614G az S-ben) trinukleotid-mutációja 231 vírusszekvenciát tartalmazó csoportot alapított (2A ábra, S-D614G klaszter), amelyek legtöbbjét Európában izolálták; 2. A C8782T (csendes) és az T28144C (L84S az ORF8-ban) dinukleotid-mutációja 208 vírusszekvenciát tartalmazó csoportot alapított (2A ábra, ORF8-L84S klaszter), amely nem monofiletikus a vizsgálatunkban (2A ábra és S3). Egy 92 szekvenciát magába foglaló különálló, monofiletikus szubkládot is meg lehet figyelni az ORF8-L84S klaszteren belül, amelyhez tartozó vírusszekvenciák legtöbbjét az amerikai Seattle-ben izolálták (2A ábra, ORF8-L84S-USA-WA-klád); 3. A G11083T (L3606F az ORF1a-ban) és a G26144T (G251V az ORF3a-ban) dinukleotid-mutációja 34 vírusszekvenciát tartalmazó csoportot alapított, amelyek többsége Hollandiából és Angliából származik. Számos kisebb monofiletikus klaszter is megfigyelhető, amelyeket különböző alapító mutációkkal lehet meghatározni (a bootstrap megerősítési érték >95). Például: 1. A G1937A (V378I az ORF1a-ban) mutáció egy 31 vírusszekvenciát tartalmazó klasztert alapít; 2. a G1440A és a G2891A mutációk az ORF1a génben, amelyek a G392D és a A876T mutációkhoz vezetnek fehérjeszinten, 12 vírusszekvenciát tartalmazó klasztert alapítanak, amelyek többsége Németországból vagy Hollandiából származik; 3. a C15325T és a C29303T mutációk az N-génben, amelyek a P344S mutációhoz vezetnek fehérjeszinten, egy 8 vírusszekvenciából álló kis klasztert alapítanak, amelyek mindegyike Kínából vagy Japánból származik. Amikor bevettük a 11 vírusizolátumot a filogenetikai elemzésbe, az egész filogenetikai térben szétszóródtak. A ZJU-1 klasztert képez az ORF1a-L3606F és ORF3a-G251V csoportokkal, mivel mindkét meghatározó mutáció megtalálható benne (2A ábra). A ZJU-2 és a ZJU-8 viszont az ORF8-L84S-sel képez klasztert, mivel mindkét alapító mutáció megtalálható bennük (2A ábra). A ZJU-9 és a ZJU-11 egy ausztrál izolátummal képez klasztert a korábban említett T22303G mutáció miatt. A csoport többi részében vagy kevés vagy új mutáció volt, így nem képeznek klasztert egyik ismert méretezhető csoporttal sem, ami a 11 mintánkkal kapcsolatban tapasztalható kifejezett diverzitással cseng egybe. Mindent összevetve néhány monofiletikus vírusklaszter nyilvánvaló földrajzi mintázatot mutat (különösen Európa és WA, USA), de ennek oka lehet az adott mutációk alapító jellege is, ami a világjárvány korai fázisában történhetett.
A betegekből származó SARS-CoV-2-izolátumok szignifikáns változatosságot mutatnak a víruskópiaszám és a citopátiás hatás tekintetében Vero-E6-sejtek fertőzésekor
Sok spekuláció és elmélet húzódik meg a vírusizolátumok szekvenálása alapján megfigyelt mutációkkal kapcsolatban. Elméletileg nem túl gyakran lehet szükség szelekciós argumentumokra ezen mutációk eredetének magyarázatához, mivel az emberről emberre terjedő fertőzési folyamat számos ismétlődő természetesen is előforduló korlátozó eseményt tartalmaz, amikor a fertőzőképes víruspopuláció akár leszűkülhet százas nagyságrendű víruskópiaszámra is (Forni et al., 2017). Ezért a jelentős mértékű genetikai diverzitást vagy a populációspecifikus történéseket e folyamat okozhatja, ahol a szelekciónak csekély szerepe van (Renzette et al., 2017). Egy korábbi szimuláció szerint (Tajima, 1989) elvégeztük a Tajima-féle semlegességi tesztet, amelyhez a vírusszekvenciák szerkesztett illesztését használtuk; a Tajima D-érték –2,8874 volt, π = 0,000641 nukleotiddiverzitással (p < 0,05), ami azt sugallja, hogy a SARS-CoV-2 genomjában a populáció közelmúltbeli expanziójának köszönhetően nagyszámú alacsony frekvenciájú allél fordul elő – ez egybecseng a vírusfertőzés alatt ismétlődően fellépő korlátozó eseményekkel. Bizonyos mutációk szelekciós előnyt vagy hátrányt jelentenek különleges körülmények között, ahogy a felfedezés is mutatja, az adaptív mutációk nagymértékben feldúsultak az S-fehérje és a humán ACE2-receptora között (Ou et al., 2020).
A betegekből származó SARS-CoV-2-izolátumokban kimutatott mutációk hatásának vizsgálatára in vitro fertőzési kísérletet végeztünk. Azért választottuk az in vitro kísérletet, mert a COVID19-betegek sokféle klinikai tünetet mutatnak a tünetmentességtől kezdve a halálig, és epidemiológiai kutatások arra utalnak, hogy a klinikai kimenetelt nagymértékben befolyásolja az egyén életkora, a komplikációk és más potenciálisan ismeretlen paraméterek (Guan et al., 2020). Először azt vizsgáltuk, vajon a vírusizolátumok sikeresen tudnak-e kötődni a Vero-E6-sejtekhez, ahogy vártuk (2B ábra), és vizuálisan azonosítottuk a virionokat a jellegzetes „koronát” kialakító S-fehérje segítségével (2C és S4A ábra). Ezután mind a 11 betegből származó vírusizolátummal fertőztük a Vero-E6-sejteket, és 1, 2, 4, 8 (kvadruplikátumokban), 24 és 48 (duplikátumokban) órával a fertőzés után begyűjtöttük a sejteket (ld. Anyagok és módszerek); összegyűjtöttük a felülúszót is, mivel a sejthalál során virionok szabadultak fel. A sejtekről 48 és 72 órával a fertőzés után DIC mikrográfokat is készítettünk, hogy a citopátiás hatásról is információhoz jussunk. Specifikus valós idejű reverz-transzkriptáz alapú polimeráz-láncreakciót (RT-PCR) használtunk az ORF1a, az E- és az N-génekre, hogy kimutassuk a SARS-CoV-2 jelenlétét (ld. Anyagok és módszerek). A ciklusküszöbértékeket (Ct) használtuk a vírusterhelés kvantifikálásához, ahol az alacsonyabb érték magasabb vírusterhelést jelent. Mivel ezek a három génnel végzett vizsgálati eredmények nagymértékben egybecsengenek (R > 0,99, p < 2,2e-16), csak az ORF1a-génnel kapcsolatos eredményeket ismertetjük (3A ábra). A negatív kontrollokból nem tudtunk szignifikáns jelet kimutatni, ezért mindegyiknek egyszerűen a 40-es Ct-értéket adtuk.
Röviden, a minták Ct-értékei az összes vírusizolátumnál alacsonyak voltak kis ingadozással 1, 2 és 4 órával a fertőzés után (3A és B ábra). Ilyen röviddel a fertőzés után a virionok még csak a sejtekhez kötődtek, hogy bejuthassanak, és a replikáció ilyenkor még ritka eseménynek számít (Schneider et al., 2012). 8 órával a fertőzés után szignifikáns csökkenést figyeltünk meg a Ct-értékekben (nőtt a vírusterhelés) a ZJU-6-os, ZJU-7-es, ZJU-9-es, ZJU-10-es, és ZJU-11-es izolátumoknál. 8 órával a fertőzés után szignifikáns csökkenést figyeltünk meg a Ct-értékekben minden vírusizolátumnál (nőtt a vírusterhelés), kivéve a ZJU-2- és ZJU-7-izolátumokat, ráadásul néhány vírusizolátumnál, nevezetesen a ZJU-10-nél és a ZJU-11-nél sokkal gyorsabb csökkenést mértünk (3A és B ábra). 48 órával a fertőzés után minden vírusizolátumnál, kivéve a ZJU-10 és ZJU-11 esetében, alacsonyabb mértékű csökkenést lehetett megfigyelni, ami alapján feltételezhető, hogy ezek már 24 órával a fertőzés után platófázisba értek (3A és B ábra). Fontos megjegyezni, hogy 24 órával a fertőzés után a ORF-8-L84S klaszterbe tartozó ZJU-2 és a ZJU-8 (az USA WA-Seattle izolátumok tartoznak ebbe a csoportba) jelentősen alacsonyabb vírusterhelést mutattak (4A ábra).
Másrészről viszont a ZJU-1-nél, amely a S-D614G kláddal (többnyire európai izolátumok) képez klasztert, a vírusterhelés 19-szerese (24,25) volt a ZJU-2 és a ZJU-8 izolátumokénak (4A ábra). Ráadásul 270-szeres különbséget (28,09) lehetett megfigyelni a vírusterhelésben a ZJU-10-es és a ZJU-2-es izolátumokat összehasonlítva 24 órával a fertőzés után. (4A és B ábra). Ezek a különbségek 48 órával a fertőzés után váltak szignifikánssá, és a megismételhetők voltak, amikor az E-génnel és az N-génnel kapcsolatos adatokat elemeztük (S4B és C ábra; S5A ábra). A különböző vírusizolátumok, amelyek mutációik alapján eltértek egymástól a genomjukban, a vírusterhelésükben szignifikáns változatosságot mutatnak a Vero-E6-sejtek fertőzésekor.
Ezután azt vizsgáltuk, hogy a magasabb vírusterhelés fokozott sejthalálhoz vezet-e (4B ábra). Amikor ezeket a sejtvonalakat mikroszkóp alatt vizsgáltuk 48 és 72 órával a fertőzés után, a citopátiás hatás (CPE) vagy sejthalál arányának szoros összefüggését találtuk a vírusterheléssel (4C ábra és S5B, C, D; 48 órával a fertőzés után, Ct vs. CPE, R = -0,72, p = 0,015), ami azt jelenti, hogy a magasabb vírusterhelés magasabb sejthalálozási aránnyal függ össze. Fontos megjegyezni, hogy a Ct-értékek negatívan korrelálnak a CPE-vel, mert az alacsonyabb Ct-értékek magasabb vírusterhelést jelentenek.

3. ábra A fertőzési kísérlet a vírusterhelés időszakos változékonyságát mutatja, amikor betegekből származó SARS-CoV-2-izolátumokkal fertőznek Vero-E6-sejteket. (A) A SARS-CoV-2 ORF1a génjére vonatkozó Ct-értékek idősoros grafikonja (a vírusterhelés többszörös inverzére vonatkozóan) a fertőzési kísérletben. Minden vírusizolátum és a negatív kontroll „C” is színesen jelölt. A p-értékeket az ANOVA módszerrel számolták, hogy összehasonlítsák mind a 11 vírusizolátum átlagait minden időpontban, kivéve a negatív kontroll „C”-t. (B) A SARS-CoV-2 ORF1a génjéhez tartozó Ct-értékek idősoros grafikonja mind a 11 betegből származó vírusizolátumra. A p-értékeket az egymást követő időpontokban t-próbával számították, és a korrigált p-értékek vannak feltüntetve.
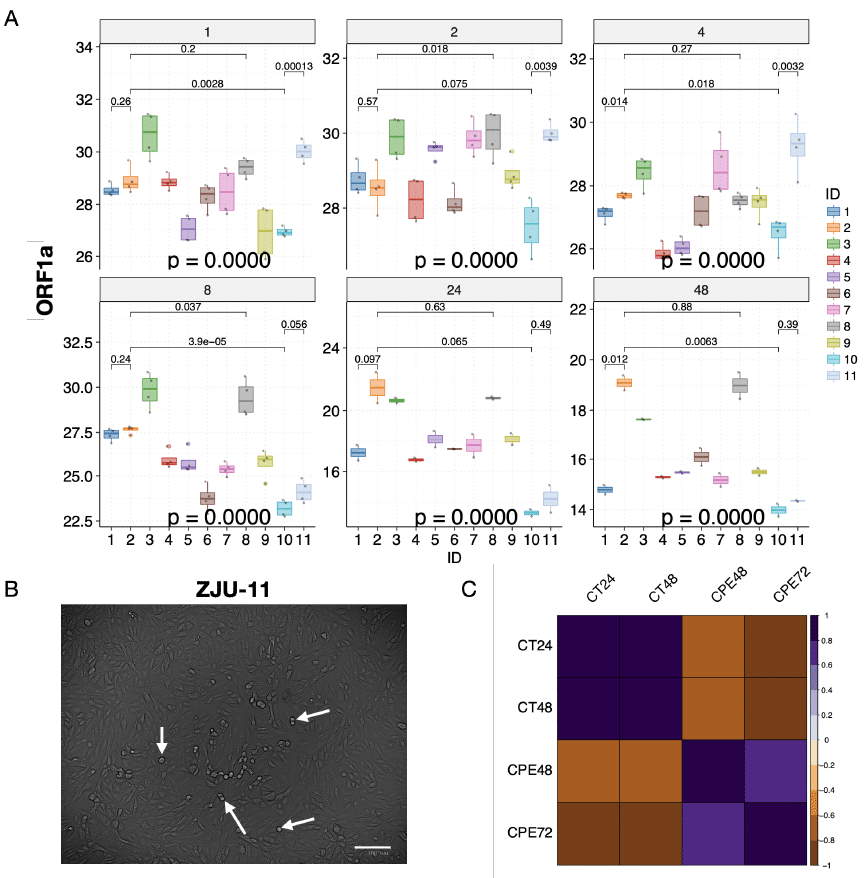
4. ábra A CPE és a vírusterhelés nagymértékben korrelált egymással. (A) A megfigyelt szignifikáns változatosság a vírusterhelésben minden egyes időpontban. A kiválasztott vírusizolátumok átlagos Ct-értékét színkódokkal jelentítettük meg. A p-értékeket az ANOVA módszerrel számoltuk, hogy összehasonlítsuk mind a 11 vírusizolátum átlagait minden időpontban. A p-értékeket az egymást követő időpontokban t-próbával számították, és a korrigált p-értékek vannak feltüntetve. (B) A citopátiás hatás 48 órával a fertőzés után látható volt a mikroszkópban, a fehér nyilak azokat a sejteket jelölik, amelyek éppen sejtlízisen mennek keresztül. (C) A citopátiás hatás (CPE) nagymértékben korrelált a vírusterheléssel (Ct) a vírusfertőzési kísérletben. Pearson-korrelációt számítottunk, és a p-értékeket eszerint korrigáltuk; csak a korrigált p-értékeket (< 0,05) tüntetjük fel. Fontos megjegyezni, hogy a Ct-értékek negatívan korreláltak a CPE-értékekkel, mivel a Ct-értékek a vírusterhelés inverzét jelentik.
Megbeszélés
A COVID19-világjárvány gyorsan terjed, és már többmilliónyian fertőződtek meg, eddig 70 000 haláleset történt világszerte. Míg sok kutatás folyik a vírus evolúciós eredetének vissszakövetésére, a fertőzés mechanizmusának kiderítésére, és a vírus ellen hatékony vakcinák és gyógyszerek fejlesztésére, a munkánk során mi a genotípus-fenotípus közötti kapcsolatot szerettük volna feltárni, amely a nagymértékű diverzitás mögött megfigyelhető, és amely a globális szekvenálási erőfeszítések eredményeként vált megfigyelhetővé (GISAID). A betegeknél tapasztalt klinikai tünetek különösen nagy változatossága miatt a genotípus-fenotípus kapcsolatát nagyon nehéz meghatározni a betegekben. Az in vitro sejtvonalak ideális rendszerként szolgálnak a különböző vírusizolátumok mutációs hatásának vizsgálatához, ha minden más zavaró tényezőt kizárunk. Habár a Vero-E6-sejtvonal nem emberi eredetű, a Vero-E6-sejtek ACE2-fehérjéje nagyon hasonló az emberéhez (S6 ábra), így közvetlen bizonyítékkal tudtunk szolgálni arra, hogy a SARS-CoV-2 képes fertőzni ezt a sejtvonalat (2B ábra).
Sok eredményt kell kiemelnünk ebből a vizsgálatból: 1. A 11 vírusizolátumban sokféle mutációt található meg, beleértve a két alapító mutációt is a két legnagyobb vírusklaszterre, amely jelenleg a világ populációját fertőzi. Ráadásul 31-ből 19 azonosított mutáció új, a viszonylag korai mintavétel ellenére, ami arra utal, hogy a vírustörzsek valós diverzitását még mindig jelentősen alulbecsülik; 2. fontos megjegyezni, hogy a T22303G és A22301C mutációk ugyanahhoz az S-fehérjében jelentkező S247R mutációhoz vezetnek (1. és S1 ábra), a létező struktúrához történt térképezés alapján kiderült, hogy ez a módosult fehérjerész az S-fehérje S1 alegységének N-terminális doménjén belüli rugalmas hurokrégióban helyezkedik el, habár az S247R pontos pozícióját eddig nem sikerült meghatározni (S7 ábra, piros ívek). Habár az N-terminális domén nem vesz részt közvetlenül az ACE2 receptorához kötődésben (Walls et al., 2020), meg kell jegyezni, hogy ez a domén közvetlenül a C-terminális domén mellett helyezkedik el, amely az ACE2 receptorát köti. Érdekes, hogy a T22303G mutációt 5 vírusizolátumban is megfigyelték, habár eltérő arányban, ami arra utal, hogy ez a specifikus mutáció már a világjárvány korai napjaiban jelen volt, és talán a vuhaniak jelentős számában is, annak ellenére, hogy a jelenlegi GISAID-gyűjteményben még mindig szinte teljesen hiányzik. Ennek oka a mutáció alapító hatása lehet, és ebben az esetben a T22303G mutáció nem terjedt el Kínán kívül a korai időszakban; 3. A trinukleotid-mutáció a ZJU-11-ben váratlan; megjegyezzük, hogy ez a specifikus vírusizolátum nagyon hatékony a vírusterhelésben és a CPE-kísérletben, és a beteg meglepő módon 45 napig pozitív maradt, és csak nemrégiben bocsátották el a kórházból (1. táblázat). E trinukleotid-mutáció funkcionális hatásának vizsgálata nagyon érdekes lenne. Megjegyezzük, hogy a jelenlegi adatbázisban egy másik trinukleotid-mutációt (G28881A, G2882A és G28883C) is azonosítottak, amely két misszensz mutációhoz vezet fehérjeszinten (S8 ábra). A cikk írásának időpontjában ez egy több mint 300 vírustörzset magába foglaló klaszterhez vezetett, ezért a mutáció vírus patogenitására gyakorolt hatását érdemes lenne vizsgálni. Végezetül, egy nemrégiben megjelent tanulmánnyal ellentétben, amelyben nem sikerült székletmintából fertőzőképes vírusizolátumhoz jutni, ebben a vizsgálatban három vírusizolátumot származott székletmintákból, ami azt jelzi, hogy a SARS-CoV-2 képes replikálódni a székletmintában (Woelfel et al., 2020).
Röviden, a tanulmányunk közvetlen bizonyítékul szolgál arra, hogy a jelenleg a SARS-CoV-2-genomban előforduló mutációk rendelkeznek azzal a potenciállal, hogy befolyásolják a vírus patogenitását. Ezért a vírus ellenőrzését sejtszinten is el kell végezni, ahol csak lehetséges, és ezen felül össze kell gyűjteni a genomszekvenálási adatokat. Továbbá az alapító mutációk jellemzése a legfőbb földrajzi alapú vírusklaszterekben nagyon hasznos lenne ahhoz, hogy meg lehessen határozni, léteznek-e olyan patogenitásbeli különbségek, amelyek segíteni tudnák a vírus ellen jelenleg is folyó harcot. Végezetül, az influenzához hasonlóan a gyógyszer- és oltóanyag-fejlesztéshez, ami nagyon sürgető, figyelembe kellene venni e felhalmozódó mutációk hatását is, különösen az alapító mutációkét, hogy elkerüljük a lehetséges csapdákat.
Ábramagyarázatok
1. táblázat A vizsgálatba bevont 11 beteggel kapcsolatos epidemiológiai információk összefoglalása. A „vírusgen.” (vírusgeneráció) az expozíciós előzmények alapján került meghatározásra.
1. ábra A 11 vírusizolátumban azonosított mutációk összegzése. A vírusgenom minden ORF-ét az NCBI NC_045512.2 jelölésmódja szerint adták meg. Ha egy mutációt egy kevert populációban figyeltek meg, akkor a két leggyakoribb allél százalékos arányait adták meg. Az aminosavszinten bekövetkező változásokat is megadták, ahol felmerült. A kék szín a közlemény írásának idején újnak számító mutációt jelöli.
2. ábra A betegekből származó SARS-CoV-2-izolátumok jellemzése. (A) 11 vírusizolátum filogenetikai analízise a GISAID-ról letöltött 725 SARS-CoV-2-szekvencia segítségével. Az 1000 bootstrap ismétléssel becsült maximum likelihood törzsfát a 11 vírusizolátum filogenetikai kontextusának szemléltetésére hoztuk létre. A nagyobb és a kisebb klasztereket színkódoztuk; a színmagyarázat az ábra bal felső részén található. Minden ZJU-mintához a zöld színt rendeltük hozzá. Az ág vastagsága a bootstrap-támogatottsággal arányos. (B) A fluoreszcensen jelölt virális S-fehérje azt mutatja meg, hogy az izolált SARS-CoV-2 virionok (zöld) a Vero-E6-sejtek külső részéhez kötődnek (a DNS kéken festődik) a bejutás előtt. Léptéksáv 50 µm. (C) Az izolált SARS-CoV-2 virionok tipikus TEM-képe, ahol a nyilak az S-fehérjékből (tüske) álló jellegzetes „koronákra” mutatnak. Léptéksáv 100 nm.
3. ábra A fertőzési kísérlet a vírusterhelés időszakos változékonyságát mutatja, amikor betegekből származó SARS-CoV-2-izolátumokkal fertőznek Vero-E6-sejteket. (A) A SARS-CoV-2 ORF1a génjére vonatkozó Ct-értékek idősoros grafikonja (a vírusterhelés többszörös inverzére vonatkozóan) a fertőzési kísérletben. Minden vírusizolátum és a negatív kontroll „C” is színesen jelölt. A p-értékeket az ANOVA módszerrel számolták, hogy összehasonlítsák mind a 11 vírusizolátum átlagait minden időpontban, kivéve a negatív kontroll „C”-t. (B) A SARS-CoV-2 ORF1a-génjéhez tartozó Ct-értékek idősoros grafikonja mind a 11 betegből származó vírusizolátumra. A p-értékeket az egymást követő időpontokban t-próbával számították, és a korrigált p-értékek vannak feltüntetve.
4. ábra A CPE és a vírusterhelés nagymértékben korrelált egymással. (A) A megfigyelt szignifikáns változatosság a vírusterhelésben minden egyes időpontban. A kiválasztott vírusizolátumok átlagos Ct-értékét színkódokkal jelentítettük meg. A p-értékeket az ANOVA módszerrel számoltuk, hogy összehasonlítsuk mind a 11 vírusizolátum átlagait minden időpontban. A p-értékeket az egymást követő időpontokban t-próbával számították, és a korrigált p-értékek vannak feltüntetve. (B) A citopátiás hatás 48 órával a fertőzés után látható volt a mikroszkópban, a fehér nyilak azokat a sejteket jelölik, amelyek éppen sejtlízisen mennek keresztül. (C) A citopátiás hatás (CPE) nagymértékben korrelált a vírusterheléssel (Ct) a vírusfertőzési kísérletben. Pearson-korrelációt számítottunk, és a p-értékeket eszerint korrigáltuk; csak a korrigált p-értékeket (< 0,05) tüntetjük fel. Fontos megjegyezni, hogy a CT-értékek negatívan korreláltak a CPE-értékekkel, mivel a Ct-értékek a vírusterhelés inverzét jelentik.
STAR Módszerek
A kísérleti modell és alanyok részletei
Az igazolt COVID19-betegek a First Affiliated Hospitalban (első számú társkórház, Kórház) kerültek felvételre 2020. január 19-e és március 5-e között. A Kórház a kínai Csöcsiang-tartomány-beli Hangzhouban található, az egyik legnagyobb tartományi kórház, amelyet a SARS-CoV-2-fertőzött betegek fogadására jelöltek ki Csöcsiang-tartományban, ezért Hangzhoun kívüli, súlyos betegeket is fogadott. 2020. január 10-ével kezdődően minden beteget, aki a kórház lázambulanciáján megjelent, klinikai ellátószemélyzet vizsgált meg SARS-CoV-2-fertőzésre a gyanús esetekre használt kritérium szerint, a kínai National Health Commission (nemzeti egészségügyi bizottság) diagnózishoz és kezeléséhez kiadott irányelvei alapján. Röviden, a betegeket a klinikai tünetek és epidemiológiai kitettségi rizikótényezőik alapján vizsgálták, figyelembe vették a korábbi Hupej-tartományba tett esetleges utazásaikat, illetve a közeli kontaktust olyanokkal, akik Hupej-tartományba látogattak a COVID19-járvány kitörése idején. A járvány terjedésével egyre inkább nőtt a Hupej-tartományon kívüli átvitel valószínűsége. Az epidemiológiai kitettség Hupej-tartományban nem volt előfeltétele annak, hogy gyanús esetnek tekintsenek valakit. Minden gyanús esetet laboratóriumi teszttel vizsgáltak, amely a SARS-CoV-2 vírusra végzett qRT-PCR vizsgálat pozitív eredményén alapult. A betegeket kizárták a vizsgálatból, ha két, 24 órás különbséggel vett minta qRT-PCR vizsgálata negatív lett. A betegek klinikai mintáit, amelyek PCR-vizsgálatból kapott Ct-értéke 28 alatt volt, összegyűjtötték a SARS-CoV-2 izolálása céljából.
Módszerek
Mintagyűjtés, vírusizolálás, sejtfertőzés és elektronmikroszkópia
Minden mintát, beleértve a köpetet, orrgaratból vett mintát és a székletet, összegyűjtöttek a COVID19-betegektől az írásos beleegyezésükkel együtt. A vizsgálatot a Csöcsiangi Egyetemi Kórház orvosi karának klinikai kutatásetikai bizottsága fogadta el (jóváhagyási engedélyszám: 2020-29) a felbukkanó fertőző betegségekre. Minden összegyűjtött mintát BSL-3-as szintű laborba küldtek a vírusizolálásra 4 órán belül.
Először a köpetet, a székletet és az orrgaratból vett mintát megfelelő térfogatban, keveréssel kezdték feldolgozni (köpet 5-10 térfogat; széklet 2 ml/100 mg; orrgaratból vett minta, 1-szeres térfogat) 2% FBS tartalmú amfotericin B-vel (100 ng/ml), penicillin G-vel (200 unit/ml), sztreptomicinnel (200 µg/ml) és TPCK-tripszinnel (4 µg/ml) kiegészített MEM-tápoldatban. A felülúszót 3000 rpm-mel, szobahőmérsékleten történő centrifugálás után összegyűjtötték. A Vero-E6-sejtek fertőzése előtt minden begyűjtött felülúszót 0,45 µm-es filteren szűrtek át, hogy eltávolítsák a sejttörmeléket stb.
A vírusfertőzés és izolálás során az átszűrt felülúszóból 3 ml-t mértek a Vero-E6-sejtekhez egy T25-ös sejttenyésztő flaskába. 35°C-on 2 órán keresztül inkubálták a sejteket, hogy kitapadjanak, majd a beoltott médiumot eltávolították, és friss tápoldatot adtak hozzá. A sejteket 35°C-on inkubálták, és naponta ellenőrizték a citopátiás hatást (CPE). A felülúszót qRT-PCR-rel tesztelték SARS-CoV-2-re (ld. lent: qRT-PCR protokoll). Amikor a qRT-PCR-vizsgálat pozitívvá vált (tipikusan 4-5 nap inkubálás után), a virionokat ultragyors centrifugálással (100 000x g 2 órán át) gyűjtötték össze a sejtkultúra felülúszójából az azt követő szekvenáláshoz, a fertőzési kísérlethez és az elektronmikroszkópos (200 kV Tecnal G2) vizsgálathoz.
Immunfluoreszcens festés
A Vero-E6-sejteket SARS-CoV-2-vel 24 órán át fertőzték, majd 80%-os (−20°C-ra hűtött) acetonban fixálták szobahőmérsékleten 10 percig. Ezután a sejteket háromszor mosták jéghideg PBS-sel, 1% BSA-val blokkolták 30 percig, és SARS-CoV-2 tüskefehérjére specifikus nyúl monoklonális antitesttel inkubálták (hígítási arány 1:200) szobahőmérsékleten 1 órán át. A sejteket ezután ismét háromszor mosták jéghideg PBS-sel, majd Alexa Fluor488®-cal konjugált kecske anti-nyúl IgG másodlagos antitesttel (Abcam, Cat No. ab150077) jelölték szobahőmérsékleten 1 órán át sötétben. A sejteket ezután ismét háromszor mosták, és 0,5 µg/ml DAPI-val (nukleáris DNS festék) inkubálták 5 percig. Az immunfluoreszcenciát egy fluoreszcens feltéttel felszerelt IX81 Olympus mikroszkóppal vizsgálták, és a képek is ezzel készültek.
Vírusfertőzési kísérlet
A Vero-E6-sejteket 24-lyukú lemezen tenyésztették, és különböző SARS-CoV-2-izolátumokkal fertőzték két párhuzamosban 0,5 MOI-val. Az inokulumokat 1 órával a fertőzés után eltávolították az 1 órás mintavételi csoportban, és 2 órával a fertőzés után a többi mintavételi csoportban. Inkubálás után a sejtkultúrákat PBS-sel mosták háromszor, és ezután 1 ml friss sejtkultúra-tápoldatot mértek minden lyukba. Ezután a sejtkultúrákat azonnal −80°C-ra fagyasztották az 1 és 2 órás csoportban, vagy tovább folytatták a tenyésztés a többi csoportban (4, 8, 24, 48 órán át), majd összegyűjtötték. Végül minden fagyasztott mintát minden időpontból egyszerre felolvasztottak, és megmérték a virális nukleinsav-tartalmat a SARS-CoV-2 qRT-PCR kittel, amely az ORF1a-, E- és N-géneket célozza (Liferiver Biotech, Shanghai). Az első két időpontban vett mintából származó eredmények a vírus sejtfelszíni megtapadási vagy bejutási képességére utalnak a célsejtekbe, míg az utolsó négy időpontban vett minta a vírusreplikáció dinamikájáról ad információt.
A citopátiás hatás (CPE) értékelése
A Vero-E6-sejtek egy rétegben nőttek, és különböző betegekből származó SARS-CoV-2-izolátumokkal fertőzték őket, ahogy azt a vírusfertőzési kísérletben leírtuk. 24, 48 és 72 órával a fertőzés után vírus indukálta citopátiás hatást lehetett megfigyelni digitális mikroszkóppal (Bio-Rad), amellyel képek is készültek. 24 órával a fertőzés után nem volt nyilvánvaló jele a CPE-nek. A 48 és 72 órával a fertőzés után készült képeket először szakértők értékelték, majd mennyiségileg meghatározták a sejthalálozási rátát.
Szekvenálási könyvtár készítése
Minden inaktivált vírusmintából a teljes RNS-t kivonták egy vírus RNS mini kit segítségével (Qiagen, Németország). A szekvenálási könyvárat a Total RNA-Seq Kittel (Kapa, Svájc) készítették el, és az Illumina Novaseq 6000 platformmal (2 x 151 bázis; Illumina Inc., San Diego, CA; BGI genomics) végezték a mélyszekvenálást.
Kvantifikálás és statisztikai elemzés
Statisztikai elemzés és vizualizálás
A statisztikai elemzés többségét és a vizualizálást az Rstudióban és R-ben végezték (verziószámok az írás időpontjában: RStudio-1.0143 és R-3.4.0), amelyhez szükség volt személyre szabott python szkriptekre (2.7.4) és shell szkriptekre (Linux) is. Az elsődleges R-csomagokat többnyire a Bioconductor project tartja karban (https://www.bioconductor.org/, az összes dependenciájával együtt). A legfontosabbak: ggplot2 (2.2.1), reshape2 (1.4.3), RColorBrewer (1.1-2), scales (0.5.0), corrplot (0.84), Hmisc (4.1-1), ggrepel (0.7.0), cluster (2.0.6), factoextra (1.0.5), plyr (1.8.4), dplyr (0.7.4), psych (1.7.8), devtools (1.13.4), ggpubr (0.1.6), tidyverse (1.2.1), gridExtra (2.3), ggsci (2.8), ggbeeswarm (0.6.0), ggpmisc (0.2.16), colorspace (1.3-2).
Általánosságban parametrikus statisztikai próbákat (t-próba, Anova, Pearson-korreláció) használtunk, ha az adateloszlás a normális eloszlásnak megfelelt (mint a qPCR méréseknél), és nem paraméteres statisztikai próbákat alkalmaztunk (Wilcoxon-próba, Kruskal-Wallis és Spearman-korreláció), ha az adathalmaz nem felelt meg a feltételezett normalitásnak. A p-értékeket a Benjamini és Hochberg (BH) módszerrel korrigáltuk (Benjamini and Yekutieli, 2001), hogy kontrolláljuk az álnegativitási találati rátát (FDR, false discovery rate), amikor többszörös összehasonlításokat vizsgáltunk, beleértve a létrehozott p-érték mátrixot, amikor korrelációs mátrixot számítottunk a különböző tulajdonságú mintáknál.
Az S-fehérje 3D-szerkezetét itt modelleztük és innen töltöttük le: https://www.rcsb.org/3d-view/6VSB/1.
A szekvenciaadatok feldolgozása, a de novo összerendezés és a mutációk azonosítása
A szekvenálási adatokat a Novaseq 6000-rel nyertük, és először az alacsony minőségű és nagyon vonalkód-kontaminált (barcode) eredményt szűrtük ki Soapnuke-kel, majd a SARS-CoV-2 (2019-nCoV) 43 teljes genomi referenciájához térképeztük BWA-MEM-mel (Li and Durbin, 2009). A SARS-CoV-2-referenciákat az NCBI-ről töltöttük le 2020. február 28-án.
Továbbá a 100 bp-nál hosszabb térképező readeket de novo összerendezéssel nyertük ki a SPAdes-szel (Bankevich et al., 2012) (v3.1.3) egy iteratív short-read genomi összerendező modullal a páros végű readekre. A K-értékek automatikusan kerültek kiválasztásra 33, 55 és 77 bp-nál ezekre a mintákra. Az összerendezés után a kontigokat blasztoltuk a nukleotid-adatbázishoz (20190301), hogy igazoljuk az eredetüket, és csak a koronavírushoz tartozó kontigokat tartottuk meg a báziskorrekcióhoz. Ezután minden mintához szűrő readeket térképeztünk vissza az eredetileg összerendezett kontigokhoz, és bam-readcountot alkalmaztunk (–min-mapping-quality=5, a többi paramétert alapértelmezettre állítottuk), hogy kiszámítsuk minden összerendezett kontigra utólag a bázisfrekvenciát. Eközben a gatk Haplotypecaller programját alkalmaztuk, hogy előhívjuk az snp/indeleket az összerendezett kontigokon, ahol a read minősége 20-nál magasabb volt. Végül a bam-fájlokat az igv-ben manuálisan vizsgáltuk meg, hogy igazoljuk minden egyes mutációt a hozzátérképezett readszám alapján, a readek közti egyensúlyt a referenciagenom pozitív és negatív szálaihoz térképeztük, és az ezeken a readeken található mutációk relatív pozíciójához.
Filogenetikai elemzés
725 jó minőségű és nagy lefedettségű SARS-CoV-2-genomot szereztünk be a GISAID-tól (letöltve 2020. március 21-én), beleértve a Yunnan RaTG13 vírustörzset és a Guangdong tobzoska vírustörzset mint külső referenciát (outgroup). A 736 genomi szekvenciát MAFFT-tal (Katoh and Standley, 2013) illesztettük egymáshoz a –thread 16 –globalpair –maxiterate 1000 opciókkal, és trimAL-lal trimmeltük a teljes hosszúságú illesztést (Capella-Gutiérrez et al., 2009) az -automated1 opcióval, hogy eltávolítsuk az illesztések hamis részeit, ami zajt okozhatott volna a filogenetikai elemzési folyamat során. Az IQ-TREE-t használtuk (Nguyen et al., 2015) a -bb 1000 -alrt 1000 -nt 64 -asr opciókkal, hogy a 736 vírusszekvencia egy 835 parszimónia informatív karaktert tartalmazó illesztéséből maximum likelihood törzsfát bootstrap eljárással becsüljünk 1000 ismétléssel. A létrehozott törzsfát iTOL-ba importáltuk és vizualizáltuk (Letunic and Bork, 2011). Elvégeztük a Tajima semlegességi tesztet, amely a vírusszekvenciák szerkesztett illesztésén alapult, a MEGA 7 használatával (Kumar et al., 2016).
Az adatok és szoftverek elérhetősége
A 11 vírusizolátum teljes genomi szekvenciáit felvittük a GISAID gyűjteménybe a következő azonosítószámokkal: EPI_ISL_415709, EPI_ISL_416042, EPI_ISL_416044, EPI_ISL_416046, EPI_ISL_415711, EPI_ISL_416047, EPI_ISL_416425, EPI_ISL_416473, EPI_ISL_416474, EPI_ISL_418990, and EPI_ISL_418991.
Kiegészítő ábrák PDF-ben letölthetőek itt (1-4) és itt (5-8).
S1 táblázat A 11 vizsgálatba bevont vírusizolátum szekvenálási statisztikájának összesítése. Fontos megjegyezni, hogy a ZJU_10 és a ZJU_11 szekvenálását külön ágon végezték.
| Azonosítószám | Nyers readek | Tiszta readek | Nyers bázisok(G) | Tiszta bázisok(G) | lefedettség | Tiszta arány | Error_rate_fq 1 | Error_rate_fq 2 |
| ZJU_1 | 239,755,367 | 227,931,734 | 71,93 | 63,96 | 2 138 916 | 88,92% | 0,04% | 0,04% |
| ZJU_2 | 211 974 282 | 195 211 677 | 63,59 | 53,28 | 1 781 761 | 83,78% | 0,04% | 0,04% |
| ZJU_3 | 421 726 717 | 378 718 257 | 126,52 | 103,91 | 3,474,902 | 82,13% | 0,04% | 0,04% |
| ZJU_4 | 485 306 221 | 434 439 201 | 145,59 | 118,99 | 3 979 199 | 81,73% | 0,04% | 0,04% |
| ZJU_5 | 232 311 525 | 205 222 721 | 69,69 | 56,11 | 1 876 400 | 80,52% | 0,04% | 0,04% |
| ZJU_6 | 342 183 998 | 273 708 578 | 102,66 | 70,92 | 2 371 668 | 69,09% | 0,04% | 0,04% |
| ZJU_7 | 227 769 540 | 191 916 976 | 68,33 | 52,18 | 1 744 975 | 76,37% | 0,04% | 0,05% |
| ZJU_8 | 355 648 629 | 331 651 060 | 106,69 | 90,7 | 3 033 140 | 85,01% | 0,04% | 0,04% |
| ZJU_9 | 287 524 792 | 260 634 803 | 86,26 | 72,7 | 2 431 194 | 84,28% | 0,04% | 0,04% |
| ZJU_10 | 136 595 606 | 101 832 137 | 40,98 | 28,24 | 944 387 | 68,91% | 0,02% | 0,03% |
| ZJU_11 | 121 791 338 | 96 729 474 | 36,54 | 27,76 | 928 335 | 75,98% | 0,02% | 0,03% |
| Átlag | 278 417 092 | 245 272 420 | 83,53 | 67,16 | 2 245 898 | 79,70% | 0,04% | 0,04% |
Szerzői hozzájárulások
M.Z., C.J., N.W., és L.L találta ki és irányította a vizsgálatot. H.Y. és X.L. végezte az összes kísérletet K.X., Y.C., L.C., F.L., Z.W., H.W., és C. Jin segítségével. C.J., M.Z., és Q.C. végezte az összes adatelemzést. C.J., Q.C., H.Y., és M.Z. írta a vázlatot, és átnézte a kéziratot a többi szerzőtől kapott visszajelzéseket figyelembe véve.
Köszönetnyilvánítás
Nagyon hálásak vagyunk Dr. X. Zhunak, Dr. L. Xiangnak, Dr. J. Jensennek és Dr. M. Lynch-nek a támogató megbeszélésekért. Köszönjük Dr. V. Billingnek a kézirat előkészítésében nyújtott segítségét.
Finanszírozás
Ezt a munkát a Zhejiang Provincial Science and Technology Department (a csöcsiangi tartományi természettudományi és műszaki intézet) legnagyobb projektje (#2020C03123), a National Science and Technology Major Project for the Control and Prevention of Major Infectious Diseases in China (a kínai nemzeti természettudományi és műszaki legfőbb projekt a legfontosabb fertőző betegségek kontrolljára és megelőzésére) (2018ZX10711001, 2018ZX10102001, 2018ZX10302206), és a Csöcsiangi Egyetem Life Sciences Institute-jának (élettudományi intézet) start-up kutatási alapja támogatta.
Irodalomjegyzék
Bai, Y., Yao, L., Wei, T., Tian, F., Jin, D.Y., Chen, L., and Wang, M. (2020). Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA – J. Am. Med. Assoc.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A.A., Dvorkin, M., Kulikov, A.S., Lesin, V.M., Nikolenko, S.I., Pham, S., Prjibelski, A.D., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477.
Bauch, C.T., Lloyd-Smith, J.O., Coffee, M.P., and Galvani, A.P. (2005). Dynamically modeling SARS and other newly emerging respiratory illnesses: Past, present, and future. Epidemiology 16, 791–801.
Benjamini, Y., and Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 11–1188.
Cao, B., Wang, Y., Wen, D., Liu, W., Wang, J., Fan, G., Ruan, L., Song, B., Cai, Y., Wei, M., et al. (2020). A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 1–13.402018ZX10711001, 2018ZX10102001, 2018ZX10302206), and start-up funds from Life Sciences Institute at Zhejiang University.
Capella-Gutiérrez, S., Silla-Martínez, J.M., and Gabaldón, T. (2009). trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics.
Capobianchi, M.R., Rueca, M., Messina, F., Giombini, E., Carletti, F., Colavita, F., Castilletti, C., Lalle, E., Bordi, L., Vairo, F., et al. (2020). Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin Microbiol Infect 0.
China National Health Committee (2020). COVID-19 clinical diagnosis and management guideline issued by National Health Commission of China, the 5th edition.
Corman, V.M., Muth, D., Niemeyer, D., and Drosten, C. (2018). Hosts and Sources of Endemic Human Coronaviruses. In Advances in Virus Research, pp. 1–188.
van Doremalen, N., Bushmaker, T., Morris, D.H., Holbrook, M.G., Gamble, A., Williamson, B.N., Tamin, A., Harcourt, J.L., Thornburg, N.J., Gerber, S.I., et al. (2020). Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N. Engl. J. Med.
Forni, D., Cagliani, R., Clerici, M., and Sironi, M. (2017). Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 25, 35–48.
Guan, W., Ni, Z., Hu, Y., Liang, W., Ou, C., He, J., Liu, L., Shan, H., Lei, C., Hui, D.S., et al. (2020).
Clinical characteristics of 2019 novel coronavirus infection in China. N. Engl. J. Med.
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., Schiergens, T.S., 41
Herrler, G., Wu, N.-H., Nitsche, A., et al. (2020). SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 1–10.
Hu, Z., Song, C., Xu, C., Jin, G., Chen, Y., Xu, X., Ma, H., Chen, W., Lin, Y., Zheng, Y., et al. (2020). Clinical characteristics of 24 asymptomatic infections with COVID-19 screened among close contacts in Nanjing, China. Sci. China Life Sci.
Katoh, K., and Standley, D.M. (2013). MAFFT Multiple Sequence Alignment Software Version 7:
Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780.
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol.
Lauer, S.A., Grantz, K.H., Bi, Q., Jones, F.K., Zheng, Q., Meredith, H.R., Azman, A.S., Reich, N.G., and Lessler, J. (2020). The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly
Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. Letunic, I., and Bork, P. (2011). Interactive Tree of Life v2: Online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760.
Lin, Q., Chiu, A.P.Y., Zhao, S., and He, D. (2018). Modeling the spread of Middle East respiratory syndrome coronavirus in Saudi Arabia. Stat. Methods Med. Res. 27, 19–1978.
Lipsitch, M., Cohen, T., Cooper, B., Robins, J.M., Ma, S., James, L., Gopalakrishna, G., Chew, S.K., Tan, C.C., Samore, M.H., et al. (2003). Transmission dynamics and control of severe acute respiratory syndrome. Science (80-. ). 300, 19–1970.
Liu, Y., Gayle, A.A., Wilder-Smith, A., and Rocklöv, J. (2020). The reproductive number of COVID-19 is higher compared to SARS coronavirus. J. Travel Med.
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., Wang, W., Song, H., Huang, B., Zhu, N., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 5–574.
Lv, L., Li, G., Chen, J., Liang, X., and Li, Y. (2020). Comparative genomic analysis revealed specific mutation pattern between human coronavirus SARS-CoV-2 and Bat-SARSr-CoV RaTG13. BioRxiv 2020.02.27.9006.
Lynch, M. (2007). The Origins of Genome Architecture. Nguyen, L.T., Schmidt, H.A., Von Haeseler, A., and Minh, B.Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol.
Ou, J., Zhou, Z., Zhang, J., Lan, W., Zhao, S., Wu, J., Seto, D., Zhang, G., and Zhang, Q. (2020). RBD mutations from circulating SARS-CoV-2 strains enhance the structure stability and infectivity of the spike protein. BioRxiv 2020.03.15.991844.
Renzette, N., Pfeifer, S.P., Matuszewski, S., Kowalik, T.F., and Jensen, J.D. (2017). On the Analysis of Intrahost and Interhost Viral Populations: Human Cytomegalovirus as a Case Study of Pitfalls and Expectations. J. Virol. 91, e01976-16.
Schneider, M., Ackermann, K., Stuart, M., Wex, C., Protzer, U., Schätzl, H.M., and Gilch, S. (2012). Severe Acute Respiratory Syndrome Coronavirus Replication Is Severely Impaired by MG132 due to Proteasome-Independent Inhibition of M-Calpain. J. Virol. 86, 10112–10122.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism.
Tortorici, M.A., and Veesler, D. (2019). Structural insights into coronavirus entry. In Advances in Virus Research, pp. 93–116.
Varia, M., Wilson, S., Sarwal, S., McGeer, A., Gournis, E., Galanis, E., and Henry, B. (2003). Investigation of a nosocomial outbreak of severe acute respiratory syndrome (SARS) in Toronto, Canada. Cmaj 1, 285–292.
Virlogeux, V., Fang, V.J., Park, M., Wu, J.T., and Cowling, B.J. (2016). Comparison of incubation period distribution of human infections with MERS-CoV in South Korea and Saudi Arabia. Sci. Rep. 6.
Walls, A.C., Park, Y.-J., Tortorici, M.A., Wall, A., McGuire, A.T., and Veesler, D. (2020). Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 44
Wang, Z., and Xu, X. (2020). scRNA-seq profiling of human testes reveals the presence of ACE2 receptor, a target for SARS-CoV-2 infection, in spermatogonia, Leydig and Sertoli cells. Viruses 1–16.
Wang, C., Li, W., Drabek, D., Okba, N.M.A., Haperen, R. van, Osterhaus, A.D.M.E., Kuppeveld, F.J.M. van, Haagmans, B.L., Grosveld, F., and Bosch, B.-J. (2020). A human monoclonal 1 antibody blocking SARS-CoV-2 infection. BioRxiv 2020.03.11.987958.
Woelfel, R., Corman, V.M., Guggemos, W., Seilmaier, M., Zange, S., Mueller, M.A., Niemeyer, D., Vollmar, P., Rothe, C., Hoelscher, M., et al. (2020). Clinical presentation and virological assessment of hospitalized cases of coronavirus disease 2019 in a travel-associated transmission cluster. MedRxiv 2020.03.05.20030502.
Wu, F., Zhao, S., Yu, B., Chen, Y.M., Wang, W., Song, Z.G., Hu, Y., Tao, Z.W., Tian, J.H., Pei, Y.Y., et al. 2020). A new coronavirus associated with human respiratory disease in China. Nature 579, 2–2.
Xiao, F., Tang, M., Zheng, X., Liu, Y., Li, X., and Shan, H. (2020). Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology.
Yu, W.-B. (2020). Decoding evolution and transmissions of novel pneumonia coronavirus (SARS-CoV-2) using the whole genomic data Comparative analyses of the chloroplast genome in carnivorous plants View project. ChinaXriv.
Zhao, Y., Zhao, Z., Wang, Y., Zhou, Y., Ma, Y., and Zuo, W. (2020). Single-cell RNA expression profiling 45of ACE2, the putative receptor of Wuhan 2019-nCov. BioRxiv 2020.01.26.919985.
Zhou, P., Yang, X. Lou, Wang, X.G., Hu, B., Zhang, L., Zhang, W., Si, H.R., Zhu, Y., Li, B., Huang, C.L., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273.